

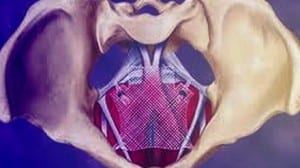
 (April 17, 2019) The US FDA announced yesterday that it was recalling all remaining surgical mesh products used on women to treat pelvic organ prolapse (POP). The FDA said it determined that Boston Scientific and Coloplast, makers of those plastic mesh products, had failed to demonstrate a reasonable assurance of their safety and effectiveness. That assurance is the premarket review standard that had applied ever since the agency reclassified POP meshes as class III (high risk) in 2016.
(April 17, 2019) The US FDA announced yesterday that it was recalling all remaining surgical mesh products used on women to treat pelvic organ prolapse (POP). The FDA said it determined that Boston Scientific and Coloplast, makers of those plastic mesh products, had failed to demonstrate a reasonable assurance of their safety and effectiveness. That assurance is the premarket review standard that had applied ever since the agency reclassified POP meshes as class III (high risk) in 2016.
That reclassification meant POP mesh makers were henceforth required to submit and obtain approval through premarket approval (PMA) applications. PMA is the FDA’s most stringent device review path for marketing medical devices in the US.
The agency said BSC and Coloplast now have 10 days to submit a plan to withdraw their products from the market.
FDA Announcement of April 16, 2019:
“The U.S. Food and Drug Administration today ordered the manufacturers of all remaining surgical mesh products indicated for the transvaginal repair of pelvic organ prolapse (POP) to stop selling and distributing their products in the U.S. immediately. The order is the latest in a series of escalating safety actions related to protecting the health of the thousands of women each year who undergo surgery transvaginally to repair POP.”
Transvaginal Mesh not FDA Approved
Plastic mesh to treat POP and SUI was never formally approved by the FDA in the usual PMA process. These meshes were, rather, “cleared” under the auspices of the agency’s 510k program, which allows some devices to reach the market if the agency determines the product in question is substantially equivalent to a product already approved. In the case of transvaginal mesh (TVM), that comparison has always been shaky at best. Polyurethane or plastic mesh was first used in hernia repair surgery in the mid 1970s, and even then it was shown to cause problems in some people. The company that made the oil-based plastic product is on record as saying it should never be implanted in the human body, but medical device companies used it anyway, and continue to use it.
FDA Dr. Jeffrey Shuren
Jeffrey Shuren, M.D., director of the FDA’s Center for Devices and Radiological Health, said, “In order for these mesh devices to stay on the market, we determined that we needed evidence that they worked better than surgery without the use of mesh to repair POP. That evidence was lacking in these premarket applications, and we couldn’t assure women that these devices were safe and effective long term.”
100,000 + TVM Lawsuits
A multi district litigation court was created to handle more than 100,000 transvaginal mesh lawsuits filed over the plastic devices used to treat POP and SUI. It was the largest ever MDL ever assembled. Jury trials resulted in several large verdicts for plaintiffs, and several thousands of cases were quietly settled for injured women. Hundreds of TVM lawsuits are still on file, and more trials are ongoing.
FDA Recalls Mesh Products for Women
One lawyer, David Matthews, who won a large jury verdict in a TVM case and later settled several others, said: “The FDA has finally done what the mesh industry would not do on its own. The adverse event reports, which reflect less than 10% of actual injuries to the women implanted with this product, coupled with multiple large jury verdicts based on expert opinions, has shown the risks of these products far outweigh their benefits, if any. This is yet more evidence that the clearance process for these dangerous products is inadequate. As lawyers who have been represented those injured by these defective products for more than eight years, we thank the FDA for declaring these plastic devices unfit for use.”
Related
- Jury awards $78 Million in Transvaginal Mesh Lawsuit
- Transvaginal Mesh Catastrophe
- Transvaginal Mesh Media Blackout
- Transvaginal Mesh
by Matthews & Associates
